
Vol. 41, n.º 1, 2008
|
REVISTA
ESPAÑOLA DE
Vol. 41, n.º 1, 2008 |
CASUÍSTICA
Axel Schmidt-Bäumler, María del Mar Martín-Jaén, Victoria Pérez-Holgado, Willy Pinto-Morales, Edmundo Juan Miralles-Sanchiz
Servicio de Anatomía Patológica del Hospital
Universitario de Valme, Sevilla.
akkisb@web.de
RESUMEN
Introducción: El síndrome hemofagocítico (SH) es un padecimiento poco frecuente, de etiología multifactorial, descrito, en sus formas secundarias, asociado a linfomas T, inmunosupresión medicamentosa, concretamente fludarabina, y a infecciones por virus, especialmente virus de Epstein-Barr (VEB). La evolución de la leucemia linfoide crónica (LLC) a linfomas más agresivos no es rara, se llama síndrome de Richter (SR), y se asocia ocasionalmente al VEB. Material y Métodos: Se realiza la autopsia de un varón de 63 años diagnosticado de LLC, estable durante años, que recae y, tras varios ciclos de quimioterapia, desarrolla una insuficiencia medular intensa que evoluciona hacia el éxitus tras fracaso multiorgánico. Resultados: El estudio macroscópico muestra datos inespecíficos. El estudio histológico revela un SH asociado a un síndrome linfoproliferativo complejo que presenta dos patrones mezclados, con predominio en ganglios y bazo del menos diferenciado, constituido por células grandes con inmunofenotipo T, y en otros órganos con persistencia del patrón linfoide de células pequeñas B consistente con el diagnóstico primitivo. El estudio inmunohistoquímico es negativo para VEB en las células tumorales. Conclusiones: La asociación de SH a Síndrome de Richter como tal no está recogida en la literatura revisada. Descartada la etiología vírica, quedan como posibles etiologías del SH el linfoma T desarrollado sobre la inicial LLC, que justifica el empeoramiento final del enfermo sin respuesta a los fármacos, o el uso de fludarabina y, subsecuentemente, insuficiencia medular final asociada al SH.
Palabras clave: Síndrome Hemofagocítico; Síndrome de Richter; Virus-Epstein-Barr; Fludarabina.
SUMMARY
Introduction: The haemophagocytic syndrome (HS) is a rare disease of multifactorial aetiology, which has been described in its secondary forms associated to T-cell lymphomas, drug induced immunsupression, concretely with Fludararabine, and in other cases to virus infection, especially with the Epstein-Barr-Virus (EBV). The evolution of chronic lymphatic leukaemia (CLL) to more aggressive lymphomas is not unusual and it is known as Richter’s Syndrome (RS), also occasionally associated to EBV. Material and methods: Autopsy of male, 63 years old, with diagnosis of CLL, stable for years, relapsing, and after various cycles of chemotherapy develops a medullar insufficiency which leads to death after multiorganic failure. Results: The macroscopic study reveals non-specific signs. The histological study reveals a HS associated to a complex lymphoproliferative syndrome with two patterns predominating in lymph nodes and spleen, the less differentiated formed of large cells with T-immunophenotype and in other organs persistence of the lymphoid pattern of small B-cells in accordance with the primitive diagnosis. The immunohistochemical study does not reveal positivity for EBV in tumour-cells. Conclusions: The association of HS to RS is not described in the revised literature. Being negative the EBV, other possible aetiologies of the HS are the T-cell lymphoma developed on the initial CLL which justifies the final worsening without drug-response or the application of Fludarabine after which the patient develops the final medullar insufficiency associated to the HS.
Key words: Hemophagocytic Syndrome; Richter’s Syndrome; Epstein-Barr-Virus; Fludarabine.
INTRODUCCIÓN
Los síndromes hemofagocíticos sistémicos son la expresión de la infiltración multivisceral por histiocitos benignos proliferados que muestran prominente hemofagocitosis. Cursan con cuadros clínicos graves de aparición brusca que con frecuencia conducen a la muerte (1). Incluyen cuadros primarios o familiares y otros secundarios o adquiridos (2) que suelen estar relacionados con infecciones (3-5), generalmente víricas por el VEB (6,7), en el curso de inmunosupresión medicamentosa (8), con linfomas (9) o leucemias agudas (5).
El síndrome de Richter es la aparición de un linfoma de alto grado de células grandes a partir de una LLC (10). En la mayoría son linfomas B (11), pero también ocurren linfomas de Hodgkin y, más raramente, linfomas T (12,13). Su desarrollo se asocia a veces a infección por el VEB (14) y, en otras ocasiones, al tratamiento utilizado contra la LLC (15,16).
Presentamos un caso autópsico de un SH desarrollado en el contexto de una LLC que evolucionó hacia un linfoma de células grandes T, por el que había recibido tratamiento inmunosupresor. No hemos encontrado casos similares de asociación de SH y SR en la literatura (Medline/Pubmed).
DESCRIPCIÓN DEL CASO
Varón de 63 años, diagnosticado de LLC en estadio «0» doce años antes, sin tratamiento inicial en los tres primeros años, que se controla después con clorambucilo y prednisona, y con ligeras variaciones hasta el año actual en el que se le extirpa un melanoma en la espalda. Seis meses antes del éxitus la LLC progresa y se trata con fludarabina, inicialmente sola y después combinada con ciclofosfamida, tratamiento que se suspende por la aparición de un cuadro de inmunosupresión profunda y compleja, con insuficiencia medular intensa, neutropenia, trombopenia y anemia, fiebre sin focalidad y cultivos de sangre y otros fluidos negativos. Fallece al cabo de un mes con fracaso multiorgánico.
En el estudio autópsico los hallazgos macroscópicos son poco relevantes, destacando adenopatías mediastínicas y abdominales, hepatomegalia de 2.250 gr con parénquima de aspecto normal, esplenomegalia de 650 gr con parénquima compacto de color azulado, y hemorragias parenquimatosas (en pulmones, riñones y testículos), serosas y mucosas (en pericardio, pleura, vejiga y mesenterio) y petequiales en la piel, que sugieren una coagulación intravascular diseminada. Además se aprecian arteriosclerosis moderada de los grandes vasos, una hipertrofia miocárdica de ventrículo izquierdo e hidrocele bilateral.
Los hallazgos histológicos muestran un síndrome hemofagocítico, con numerosos macrófagos cargados de hematíes y otras células sanguíneas enteras o fragmentadas, que ocupan (e incluso borran focalmente) la arquitectura del bazo, los ganglios linfáticos (fig. 1) y la médula ósea, y también, aunque con menor intensidad, los sinusoides hepáticos (fig. 2) y los capilares pulmonares. Además, la morfología ganglionar y esplénica se encuentra borrada por una proliferación linfocitaria polimorfa con presencia de células de tipo inmunoblástico, algunas binucleadas sternbergoides, y con mitosis típicas y atípicas (fig. 3). También se observan agregados de células linfoides pequeñas y zonas de aspecto granulomatoso con despoblación linfocitaria y proliferación de células epitelioides. En el esófago y la médula ósea existen focos pleomórficos y en el hígado, focos linfocitarios pequeños. El cuadro hemorrágico macroscópico parece corresponderse con el diagnóstico sugerido. Otra alteración histológica no relacionada con el cuadro principal es una atrofia tubular testicular.
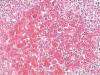
Fig. 1: Ganglio
linfático con extensa eritro- y linfofagocitosis (HE x400).
Fig. 2: Parénquima
hepático con hemofagocitosis sinusoidal. (HE x200).
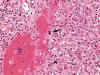
Fig. 3: Proliferación
linfoide polimorfa con signos de mitosis (flecha) y células sternbergoides
(cabeza de flecha) en ganglio linfático mediastínico (HE x400).
El estudio inmunohistoquímico realizado con los métodos convencionales (inmunotinción automatizada con estreptavidina biotina, procesador Autostainer y reactivos de Dakocytomation) pone de manifiesto la naturaleza T de las células polimorfas, al expresar CD 3 y UCHL-1 (fig. 4), y B de las linfoides pequeñas, que se definen con CD 20, CD 79 alfa y bcl-2 (fig. 5), lo que parece confirmar la existencia de un Síndrome de Richter. Las células fagocíticas son CD 68 positivas y no se demuestra positividad en ningún tipo con LMP-1 para VEB.

Fig. 4: Proliferación
linfoide polimorfa en ganglio linfático con marcadores inmunohistoquímicos
positivos para proliferación de células T; A: CD 3 (x200); B: UCHL-1 (CD 45Ro)
(x400).

Fig. 5: Proliferación
linfoide polimorfa en ganglio linfático con marcadores inmunohistoquímicos
positivos para proliferación de células B; A: CD 79 alfa; B: CD 20; C: bcl-2
(x200).
Se sospechan como enfermedades fundamentales y probables causas del éxitus un SR compuesto por LLC B y Linfoma T periférico asociado a SH, diagnóstico confirmado por el estudio realizado en el Centro Nacional de Investigación Oncológica, Madrid (CNIO), donde se mandó el caso como consulta, confirma nuestra sospecha inicial y reafirma el diagnóstico.

DISCUSIÓN
Los síndromes hemofagocíticos sistémicos incluyen formas familiares primarias, que aparecen en la niñez, y formas secundarias, que se asocian a infecciones o a procesos malignos y afectan a un mayor rango de edades (2). En todos los tipos de SH, la activación macrofágica ocurre por aumento de niveles de citoquinas liberadas por macrófagos o células T (1). Las formas secundarias a infección tienen lugar en pacientes con una inmunodeficiencia subyacente, con frecuencia secundaria a tratamiento inmunosupresor y por el VEB (6,7), aunque ocasionalmente pueden desencadenarse por cualquier tipo de infección vírica (3-5). Las formas asociadas a neoplasias pueden aparecer como consecuencia de la inmunosupresión secundaria al tratamiento de la neoplasia, pero también se ven en neoplasias de células T o de células Natural Killer (NK) (6,7,9).
El espectro clínico es variable, pero los pacientes presentan rasgos de enfermedad grave con fiebre alta y síntomas constitucionales. Muestran hepatoesplenomegalia y, a veces, adenopatías. Los datos analíticos asociados revelan una coagulopatía con rasgos de coagulación intravascular diseminada, alteraciones en la función hepática y aumento de los niveles de triglicéridos (1). El examen medular presenta citopenias de dos o tres líneas, con variable densidad celular en el cilindro y con hiperplasia histiocítica y prominente hemofagocitosis: Abundantes histiocitos de aspecto citológicamente benigno muestran sus citoplasmas ocupados por células hematopoyéticas maduras e inmaduras, vacuolas y gránulos. Este carácter maduro y sin atipia de los histiocitos diferencia el cuadro de otros procesos histiocíticos malignos. El estudio inmunohistoquímico con CD 68 define la población proliferada. El anticuerpo LMP-1 puede demostrar la existencia de VEB. La infiltración multivisceral es la que condiciona la clínica y en ocasiones lleva al éxitus (1,6).
El síndrome de Richter o transformación inmunoblástica de una LLC se caracteriza por el desarrollo de un linfoma de células grandes, enfermedad de Hodgkin o leucemia de células blásticas a partir de la LLC. Ocurre en un 5% de las LLC (10). Generalmente son linfomas B (11) y excepcionalmente T (12). Puede quedar evidencia de la inicial LLC en áreas separadas (10). En muchos casos ocurre transformación del clon original, pero ocasionalmente hay una evolución aparente de un nuevo clon neoplásico (13). Esta transformación hacia linfomas más agresivos se ha descrito asociada a infección por VEB (14) y también al uso de determinados inmunosupresores como fludarabina (15,16). Cursa con un cambio brusco en la evolución clínica, aparición de fiebre, pérdida de peso, adenopatías localizadas y disglobulinemia (10).
En nuestro caso, la transformación a linfoma T de alto grado se demuestra en la autopsia, pero produce cambios clínicos seis meses antes del éxitus, quizás enmascarados por la superposición de la sintomatología atribuible al SH. Morfológicamente queda poca expresión residual de la LLC, predominando las alteraciones hemofagocíticas y del linfoma T. El caso reúne gran parte del espectro descrito, tanto clínico como morfológico, de los síndromes hemofagocíticos. No hemos podido demostrar anticuerpos contra el VEB ni otras infecciones oportunistas favorecidas por la inmunosupresión, y que se describen como desencadenantes del cuadro. Sí existió un tratamiento variable con inmunosupresores y, tras uno de los cambios, es cuando parece ocurrir la crisis clínica inicial del SH. Estas modificaciones últimas en los tratamientos están relacionadas con la mala evolución de la LLC. El éxitus puede justificarse tanto por el SH como por el linfoma.
La asociación de SH a Síndrome de Richter como tal no está recogida en la literatura publicada en Medline. Descartada la etiología infecciosa, sobretodo por el VEB, quedan como posibles etiologías del SH el linfoma T desarrollado sobre la inicial LLC, que es el tipo de linfoma que se asocia más frecuentemente con SH. Como otra posibilidad no desdeñable o quizás potenciadora de la anterior, hay que tener en cuenta el estado de inmunosupresión terapéutico, que por sí mismo puede producirlo, producir un SH aunque con mayor frecuencia se descrie como substrato sobre el que diferentes agentes infecciosos pueden desencadenarlo, fenómeno no demostrado en nuestro caso.
BIBLIOGRAFÍA
Foucar K. Bone marrow pathology. 2nd Ed. Chicago. ASCP Press. 2001, 526-8.
Kumakura S. Hemophagocytic syndrome. Editorial. Int Med 2005; 44: 278-80.
Ruiz-Arguelles GJ, Arizpe-Bravo D, Garces- Eisele J, Sanchez-Sosa S, Ruiz-Arguelles A, Ponce de Leon S. Tuberculosis-associated fatal hemophagocytic syndrome in a patient with lymphoma treated with fludarabine. Leuk Lymphoma 1998; 28: 599-602.
Gupta P, Hurley RW, Helseth PH, Goodman JL, Hammerschmidt DE. Pancytopenia due to hemophagocytic syndrome as the presenting manifestation of babesiosis. Am J Hematol 1995; 50: 60-2.
Rao RD, Morice WG, Phyliky RL. Hemophagocytosis in a patient with chronic lymphocytic leukemia and histoplasmosis. Mayo Clin Proc 2002; 77: 287-90.
George TI, Jeng M, Berquist W, Cherry AM, Link MP, Arber DA. Epstein-Barr-virus-associated peripheral T-cell lymphoma and hemophagocytic syndrome arising after live transplantation: Case report and review of the literature. Pediatr Blood Cancer 2005; 44: 270-6.
Suzumiya J, Ohshima K, Kanda M, Kumagawa M, Nagano M, Hirata M, et al.. Epstein-Barr virus (EBV) – induced B-cell proliferative disorder after chemotherapy in a patient with hemophagocytic lymphohistiocytosis with associated EBV-induced T-cell proliferation. Leuk Lymphoma 2000; 37: 593-604.
Terrovitis JV, Matsouka C, Anagnostopoulos A, Anastasiou-Nana MI, Dimopoulos AM. Hemophagocytic lymphohistiocytosis after chemotherapy for multiple myeloma. Clin Lymphoma 2004; 5: 194-6.
Domman-Scherrer C, Zimmermann D, Hassam S, Odermatt BF, Risti B, Maurer R. T-cell rich B-cell lymphoma associated with hemophagocytic syndrome. Verh Dtsch Ges Pathol 1992; 76: 122-5.
Tsimberidou AM, Keating MJ. Richter syndrome. Biology, incidence, and therapeutic strategies. Cancer 2005; 103: 216-28.
Nakamura N, Abe M. Richter syndrome in B-cell chronic lymphocytic leukemia. Pathol Int 2003; 53: 195-203.
Novogrudsky A, Amorosi EL, Gottesman SR. High-grade T-cell lymphoma complicating B-cell chronic lymphocytic leukemia: an unusual manifestation of «Richter’s syndrome». Am J Hematol 2001; 66: 203-6.
Martinez A, Pittaluga S, Villamor N, Colomer D, Rozman M, Raffeld M, et al.. Clonal T-cell populations and increased risk for cytotoxic T-cell lymphomas in B-CLL patients. Clinico-pathologic observations and molecular analysis. Am J Surg Pathol 2004; 28: 849-58.
Ansell SM, Li CY, Lloyd RN, Phyliky RL. Epstein- Barr virus infection in Richter’s transformation. Am J Hematol 1999; 60: 99-104.
Keating MJ, O’Brien S, Lerner S, Koller C, Beran M, Robertson LE, et al. Lon-term follow-up of patients with chronic lymphocytic leukemia (CLL) receiving Fludarabine regimen as initial therapy. Blood 1998; 92: 1165-71.
Keating MJ, O’Brien S, Albitar M, Lerner S, Plunkett W, Giles F et al. Early results of a chemoimmunotherapy regimen of Fludarabine, Cyclophosphamide, and Rituximab as initial therapy for chronic lymphocytic leukemia. J Clin Oncol 2005; 23: 4079-88.
![]()