
Vol. 35, n.º 1, 2002
| REVISTA
ESPAÑOLA DE Vol. 35, n.º 1, 2002 |
Aurelio Ariza
Servicio de Anatomía Patológica, Hospital Germans Trias i Pujol. Universidad Autónoma de Barcelona
INTRODUCCIÓN
Habida cuenta que las encefalopatías espongiformes transmisibles están causadas por unos agentes infecciosos denominados priones, cabe comentar en primer lugar en qué consisten estas pequeñas proteínas. La familiaridad, a menudo rayana en la temeridad, que los patólogos han desarrollado en relación con los virus, micoplasmas, rickettsias, clamidias, bacterias, protozoos y gusanos se desvanece ante el prión, cuyo concepto mismo es tan heterodoxo que la comunidad científica lo ha contemplado con grandes dosis de escepticismo hasta fecha muy reciente (1-4).
A diferencia de los agentes infecciosos convencionales, que en general son ajenos al organismo humano, los priones forman parte normal del mismo. De hecho, los humanos comparten estas proteínas con el resto de la escala evolutiva. A pesar de esta conservación tan amplia, se desconoce la función de los priones y la razón de su presencia en la superficie de tipos celulares tan diversos como los espermatozoides y las células reticulares dendríticas. Las concentraciones máximas de priones, sin embargo, se dan en las neuronas, hecho que concuerda con que las enfermedades priónicas hasta ahora reconocidas estén relacionadas con el sistema nervioso (5,6).
Los problemas surgen cuando el plegamiento normal del prión se modifica y la proteína adquiere una conformación anómala. Al imaginar la cadena de aminoácidos (estructura primaria) del prión, se ha de tener en cuenta que esta cadena se pliega sobre sí misma (estructura secundaria) y aún torna a plegarse de manera más compleja hasta adquirir una forma globular (estructura terciaria). La conformación de ciertas partes de esta estructura globular es muy característica, ya que en ellas la proteína se dispone a manera de tirabuzón (dominio en hélice alfa) o a manera de hoja de papel con múltiples dobleces paralelos (dominio en lámina beta) (2,5,6).
En el prión normal predominan los dominios en hélice alfa sobre los dominios en lámina beta. Los cambios conformacionales que confieren patogenicidad a los priones consisten precisamente en la adquisición de dominios en lámina beta, que llegan a predominar sobre los dominios en hélice alfa (2,5,6). Tal riqueza de dominios en lámina beta es responsable de que los agregados de priones anómalos posean las características tintoriales de la amiloide, con las que los patólogos están muy familiarizados. De hecho, el rojo Congo se ha propuesto como posible tratamiento de las enfermedades priónicas, pues al unirse a los priones anómalos alteraría la estructura de los mismos y los inactivaría (7,8).
En cualquier caso, las neuronas, que tan eficientes son en la síntesis y degradación de los priones normales, resultan ser completamente incapaces de deshacerse de los priones anómalos, cuyos agregados, mediante mecanismos aún desconocidos, provocan la muerte neuronal, intensa gliosis reactiva y aparición de vacuolas en el tejido cerebral (2). Del aspecto espongiforme que las vacuolas imprimen al parénquima encefálico deriva parte de la designación de este conjunto de enfermedades. La alusión a otro rasgo prominente, la transmisibilidad, completa la denominación convencional de estos procesos: encefalopatías espongiformes transmisibles (EET).
La siguiente pregunta que surge es cómo un prión normal puede cambiar su plegamiento y convertirse en un prión anómalo. Contemplado de forma simplificada, el cambio inicial puede tener lugar espontáneamente o producirse por la influencia de priones anómalos que han sido introducidos en el organismo bien mediante ingestión de productos contaminados o bien de forma yatrógena. Los priones anómalos, al ponerse en contacto con los priones normales, hacen que éstos pierdan su plegamiento habitual y adopten una conformación anómala. De esta manera se va extendiendo de forma lenta y silente el proceso hasta que muchos años después, destruido ya un gran número de neuronas, aparecen los primeros síntomas (1-3,5-8).
En contraposición a los virus y bacterias, que tienden a replicarse con rapidez y dejan sentir sus efectos en breve plazo, los priones anómalos son lentos en su propagación y los síntomas de las enfermedades priónicas pueden tardar en aparecer entre 10 y 40 años. Esta imprecisión en el conocimiento del período de incubación de las EET hace que las predicciones del número de casos humanos que pueden originarse a partir de la epidemia de la vacas locas oscilen ampliamente entre unos pocos centenares y varios millones (4,9,10).
Pero la conexión entre vacuolas, transmisibilidad y priones no siempre ha sido tan evidente. El laborioso esclarecimiento de este enlace, posibilitado por disciplinas tan dispares como la neuropatología, la antropología y la biología molecular y jalonado por dos premios Nobel, se remonta al menos al siglo XVIII y ha tenido escenarios tan diversos como las islas del Pacífico Sur y las praderas inglesas. La historia de las EET, además de interesante, es imprescindible para entender unas enfermedades que de pronto han dejado de ser un arcano para expertos y se han convertido en una obsesión colectiva que demanda insistentemente la atención de los patólogos.
LAS EET ENTENDIDAS DESDE SU HISTORIA
Tembladera: el sacrificio de los corderos
La historia de las EET comienza en las primeras décadas del siglo XVIII, cuando en varios países europeos aparecen descripciones de una enfermedad de las ovejas conocida como tembladera en castellano, tremblante en francés y scrapie en inglés. Las designaciones en castellano y francés aluden al temblor como manifestación importante de la enfermedad, en tanto que el término inglés scrapie delata la tendencia que tienen los animales enfermos a rascarse contra las estacas y otros objetos para aliviar su prurito. Las ovejas afectadas se vuelven irritables y temblorosas, luego desarrollan prurito, convulsiones, parálisis y ceguera y mueren al cabo de unos meses.
La tembladera, cuya amplia diseminación entre las ovejas mereció que fuese discutida en el Parlamento británico en el siglo XVIII, sigue siendo frecuente en nuestros días. Afortunadamente, no hay pruebas de que la enfermedad se contagie a los humanos mediante el consumo de cordero, como bien parecían saber ya hace dos siglos y medio algunos autores alemanes, que aconsejaban destinar la carne al consumo de los sirvientes tras el sacrificio de los corderos enfermos. Sin embargo, a la vista de la transmisibilidad oral de la encefalopatía espongiforme bovina (EEB) a las ovejas en un contexto experimental, cabe preguntarse si algunos casos de ovejas con EET están producidos por el agente de la EEB (y no por el agente del scrapie), lo que plantearía un riesgo para el consumo humano. El scrapie, que actualmente es en muchos países un proceso enzoótico (término equivalente a endémico en patología humana), afecta al ganado ovino del Reino Unido en una proporción que oscila entre el 0,5 y el 1% (3,7,8,11).
Creutzfeldt y Jakob: dos nombres para una demencia
El segundo capítulo en la historia de las EET se inicia en Alemania en 1920, cuando Creutzfeldt (12) y más tarde Jakob (13-15) describen uno y cinco casos, respectivamente, de la que con posterioridad se conocería como enfermedad de Creutzfeldt-Jakob (ECJ), si bien luego se ha determinado que ni el paciente de Creutzfeldt ni dos de los de Jakob padecían lo que hoy se entiende por ECJ (16). La ECJ es la EET humana más frecuente, con una incidencia de un caso por cada millón de habitantes y año. De las diversas formas de la ECJ, la más común es la esporádica, que es responsable del 85% de los casos, en tanto que las formas yatrógenas y familiares (véase más adelante) se reparten el resto. La forma esporádica de la ECJ suele afectar a pacientes de unos 60 años (es muy rara por debajo de los 30 o por encima de los 80) y se caracteriza por la instauración sutil de depresión y problemas de memoria que en el plazo de unos 6 meses progresan a demencia, mioclonias y la muerte (16).
Kuru: el ágape de los caníbales
El tercer capítulo de esta historia discurre en Papúa-Nueva Guinea, en el Pacífico Sur, adonde se desplaza el norteamericano Carleton Gajdusek en 1950. Papúa-Nueva Guinea, donde cada una de las numerosas tribus que la pueblan posee una lengua que es totalmente incomprensible en los valles vecinos, es una de las regiones con más diversidad lingüística del mundo (tan sólo la aventajan California y el Cáucaso). No es de extrañar, por tanto, que el progreso en este capítulo del conocimiento de las EET se debiese en gran parte a los dilatados conocimientos de antropología y lingüística que Gajdusek suma a su arsenal de pediatra y virólogo. Asimismo, es insoslayable la crucial aportación de Vicent Zigas, cuyo libro «Laughing Death — The Untold Story of Kuru» (17) es el gratificante escenario de un raro y feliz encuentro entre ciencia y literatura.
En la tribu de los Fore, Gajdusek se encontró con una enfermedad neurológica que hacía estragos entre las mujeres y los niños. Los Fore llamaban a la enfermedad «kuru», que en su lengua quiere decir temblor, y estaban muy familiarizados con el curso de la misma. Sabían que los temblores iniciales se seguían inexorablemente de inestabilidad de la marcha, alteraciones del lenguaje, risa inmotivada, coma y, al cabo de unos 16 meses, la muerte. Gajdusek, que sospechó estar ante una enfermedad epidémica relacionada de alguna manera con los hábitos alimentarios de la tribu, observó que si bien los hombres añadían algunas piezas de caza menor a su dieta básica de judías y batatas, las mujeres y los niños carecían de este suplemento proteico. Como alternativa, las mujeres habían recurrido a un rito muy peculiar, consistente en practicar el canibalismo con los difuntos de la familia. Aunque los Fore consideraban a los leprosos como no aptos para ser objeto de dicho consumo ritual, los enfermos de kuru les parecían limpios y exentos de riesgo.
La conexión entre canibalismo y kuru estaba clara para Gajdusek, pero le importunaban la ausencia de signos clínicos de infección en los enfermos y la negatividad repetida de los cultivos de líquido cefalorraquídeo. En un intento por desvelar el enigma, envió muestras autópsicas de cerebro a Maryland, a sus antiguos compañeros de los National Institutes of Health, que, sorprendentemente, detectaron grandes semejanzas entre los hallazgos neuropatológicos del kuru y los de la ECJ. Además de compartir vacuolas, pérdida neuronal y gliosis, ambas enfermedades presentaban un curso clínico parecido. Pero la constatación de estas similitudes complicó aun más la cuestión, al plantear cómo es que una epidemia de una tribu del Pacífico podía presentar unos hallazgos neuropatológicos semejantes a los de una enfermedad sumamente rara que afectaba al azar tan sólo a una persona por millón y año en el resto del mundo (17,18).
Gajdusek: la transmisión a los chimpancés
El siguiente avance importante en el conocimiento de las EET se produce cuando el inglés William Hadlow, experto en tembladera, lee algunas de las descripciones de kuru hechas por Gajdusek y se apercibe de las similitudes neuropatológicas y sintomáticas entre éste y la tembladera. En una carta a The Lancet, Hadlow (19) establece el paralelismo entre el kuru y la tembladera y apunta hacia una de las líneas principales de la investigación en este campo. Según Hadlow, puesto que, tal como habían demostrado ya Cuillé y Chelle en 1936 (20), las ovejas sanas desarrollan tembladera si se les inyecta intracerebralmente tejido encefálico procedente de ovejas enfermas, cabía preguntarse qué ocurriría al inyectar a animales sanos tejido encefálico de personas con kuru. En respuesta, en 1966 Gajdusek y Gibbs (21) inyectaron homogeneizados de cerebro de enfermas con kuru a chimpancés, que desarrollaron los síntomas y los cambios neuropatológicos de la enfermedad. Con ello quedó constatada la transmisibilidad del kuru. A continuación se comprobó que la inyección a chimpancés de homogeneizados de cerebros de enfermos con ECJ también transmitía la enfermedad (22).
La demostración de la transmisibilidad del kuru y la ECJ trajo consigo la concesión del Premio Nobel a Gajdusek en 1976 y puso fin al canibalismo entre los Fore. Con ello prácticamente desapareció el kuru, pero permaneció la incógnita sobre la naturaleza del agente infeccioso implicado. En años posteriores se fueron identificando casos espontáneos de EET en otras especies animales como el visón (1965), el alce (1980) y el ciervo (1980), pero el agente responsable continuó siendo un misterio que se pretendió resolver invocando el papel de los denominados virus lentos o no convencionales.
Prusiner: la revolución de los priones
Es en la década de los 80 cuando Stanley B. Prusiner entra en escena para derrocar la teoría de los virus lentos y, tras un largo pulso contra el escepticismo generalizado, convertir su hipótesis de los priones en la explicación canónica de la etiopatogenia de las EET. Prusiner consiguió la purificación bioquímica del agente del scrapie y acuñó el acrónimo prión (partícula proteinácea infecciosa) para designar a los agentes responsables de las EET, que consistirían únicamente en proteína y carecerían de ácidos nucléicos. Para agilizar la terminología, Prusiner denominó PrP (de proteína priónica) al prión en general, PrPC al prión celular normal y PrPSc al prión patológico (en alusión a su relación con el scrapie).
Las ideas de Prusiner adquirieron fuerza con la clonación del gen responsable de la síntesis de los priones y la imposibilidad de transmitir la enfermedad a ratones knock-out desprovistos de dicho gen. Finalmente, el paradigma de los priones recibió el espaldarazo de la comunidad científica al ser galardonado Prusiner con el Premio Nobel en 1997 (5,6,23,24).
Como antecedentes del trabajo de Prusiner cabe resaltar las aportaciones de Tikva Alper, que sugirió que el agente infeccioso podría ser una proteína, y de JS Griffith (25), que fue el primero en proponer la hipótesis de «sólo proteína» (protein-only hypothesis) (7).
Formas yatrógenas: contaminación de hormonas, meninges y córneas
Entre tanto se estaban produciendo otros acontecimientos que hacían aun más patente la complejidad de este grupo de enfermedades. A partir de la década de los 60 del siglo XX, miles de niños con problemas de enanismo recibieron inyecciones de hormona de crecimiento procedente de hipófisis de cadáveres humanos (hGH). Este tipo de tratamiento fue interrumpido en 1985, ante la aparición en estos pacientes de una forma peculiar de ECJ cuya presentación tenía lugar ya en la tercera década, y no a partir de los 50 años, como es propio de la forma esporádica de ECJ. Entre las 27.000 personas que, a nivel mundial, habían recibido hGH antes de que se prohibiese su administración en 1985, se han registrado hasta la fecha 22 casos de ECJ yatrógena en Estados Unidos. Otras formas yatrógenas de ECJ, descritas con anterioridad, son las asociadas a injertos de duramadre, trasplante de córnea e implantación de electrodos en el cerebro (26,27).
Vacas locas: la caída de la barrera entre especies
Mientras que Stanley Prusiner defendía su hipótesis sobre los priones y la comunidad médica descubría la forma yatrógena de ECJ asociada a hGH, en el Reino Unido los ganaderos y veterinarios eran testigos de la aparición de una nueva enfermedad del ganado vacuno. Esta pasó a denominarse encefalopatía espongiforme bovina (EEB, o enfermedad de las vacas locas) al comprobarse que sus hallazgos neuropatológicos son semejantes a los de la tembladera, el kuru y la ECJ (28). El primer caso oficial de EEB en el Reino Unido, la denominada «vaca 133», data de 1985.
Independientemente de si la EEB se originó espontáneamente en las vacas o si éstas comenzaron a sufrirla al ser alimentadas con despojos de ovejas con scrapie, no cabe duda que el brote de EEB ha sido favorecido por la práctica de alimentar a las vacas con despojos de otras vacas. Puesto que la EEB parecía asociarse al consumo de ovejas o vacas enfermas, tres años más tarde el gobierno británico prohibió el uso de despojos de oveja, vaca y otros rumiantes en la fabricación de piensos para vacas británicas. No se dieron instrucciones, en cambio, sobre el uso de dichas harinas cárnicas en la alimentación de otros animales, tales como gallinas, cerdos y conejos, ni se prohibió la exportación de las mismas.
A medida que la enfermedad se fue extendiendo a gatos domésticos (1990) y monos en zoológicos (1992) se fue comprendiendo que la barrera entre especies no era tan impermeable como hasta entonces se había creído y fue generalizándose el pánico entre la población británica, a la que ya en 1990 se había aconsejado abstenerse del consumo de las partes más infecciosas de la vaca (encéfalo, medula espinal, bazo, amígdalas, timo e intestinos) (1,24,28).
Nueva variante: la amenaza de una nueva plaga
Los peores temores se vieron confirmados cuando, en 1995, las autopsias de varios jóvenes con cuadros clínicos reminiscentes de ECJ pusieron de manifiesto lo que parecía ser la forma humana de la EEB, también conocida como variante de la enfermedad de Creutzfeldt-Jakob (vECJ). La vECJ afecta a sujetos más jóvenes (entre 16 y 48 años) que la forma esporádica y se caracteriza por parestesias, pérdida de peso, fatigabilidad, ataxia, pérdida de memoria, depresión, insomnio, alteraciones del comportamiento, coma y la muerte entre los 9 y los 39 meses después del inicio. Las alteraciones electroencefalográficas y la elevación de la proteína 14.3.3, acompañantes habituales de la forma esporádica de la ECJ, suelen estar ausentes en la vECJ (29,30).
Cuando en 1996, una década después de la identificación de la primera vaca loca, el gobierno británico al fin prohibió la fabricación de harinas cárnicas y la exportación de las mismas, ya se habían distribuido millones de toneladas por más de cien países, sobre todo de Europa del Este, Oriente Medio y Asia. Las consecuencias de tan desafortunada gestión de la crisis están por comprobar, pero la desconfianza generada ha puesto bajo sospecha cualquier producto derivado de las vacas, desde las cremas para la piel hasta las comidas para los niños. Y aunque no hay ninguna evidencia que avale la capacidad de los productos de la sangre para transmitir las EET, son ya varios los países que rechazan a los donantes procedentes del Reino Unido. El pánico social, sin embargo, no está muy justificado si se tiene en cuenta que en el Reino Unido el consumo indiscriminado de miles de vacas enfermas tan sólo ha provocado alrededor de 90 casos de vECJ. Como contrapunto, hay autores que claman que los casos de vECJ habidos hasta la fecha podrían corresponder a las personas que sufrieron la exposición durante la fase preclínica de la EEB y que podrían producirse muchísimos más casos como cosecuencia de exposiciones más tardías (30).
En última instancia, está claro que esta crisis obliga a poner orden en la industria alimentaria, pero el bajo riesgo de que las personas contraigan la enfermedad no justifica la alarma social que desde el año 2000 se ha apoderado de la Europa continental y que, desde la detección del primer caso español de EEB (la «vaca Parrula»), se ha extendido en nuestro país. Es de esperar que la interrupción del canibalismo animal tenga a la postre unos efectos tan positivos sobre la EEB y la vECJ como los tuvo la prohibición del canibalismo humano sobre el kuru (10,30).
Formas familiares: mutaciones en el cromosoma 20
Junto al kuru y las diversas formas de ECJ (esporádica, yatrógena, variante, familiar), el síndrome de Gerstmann-Sträussler-Scheinker (GSS) y el insomnio familiar fatal (IFF) completan el panorama actual de las EET humanas (tabla I). El GSS, descrito en 1936 (31), se caracteriza por dificultades en la marcha, el lenguaje y la deglución, demencia y la muerte tras un curso de 2 a 6 años, en tanto que en el IFF, descrito en 1986 (32), el insomnio progresivo se sigue de ataques de pánico, fobias, alucinaciones, demencia y, tras unos 18 meses, la muerte.

El GSS y el IFF comparten con la forma familiar de la ECJ el hecho de que la conformación anómala del prión está provocada no por modificaciones postraduccionales de la proteína sino por mutaciones en el gen de los priones, que se aloja en el cromosoma 20. Tales mutaciones resultan en la substitución de aminoácidos que, por su posición estratégica, influencian la forma de plegamiento del prión, que en consecuencia es sintetizado ya con una conformación anómala (33). Por ejemplo, la anomalía genética asociada con el GSS es la sustitución de una prolina por una leucina en el codón 102 del gen de los priones. Cuando se logró la transmisión experimental del GSS se provocó un gran desconcierto, ante la paradoja aparente de que una enfermedad genética fuese al mismo tiempo transmisible (2). Alcanzado este punto, conviene hacer una incursión en los aspectos moleculares de los priones.
BIOLOGÍA DE LOS PRIONES
La proteína priónica
Como se ha señalado más arriba, Stanley Prusiner purificó la proteína patológica (PrPSc) y postuló que la replicación de los priones ocurre mediante una reacción en cadena según la cual PrPSc recluta PrPC y lo convierte en nuevo PrPSc (5,6,23,24) (fig. 1). Para algunos autores, esta interacción entre PrPC y PrPSc necesitaría del concurso de una tercera proteína (proteína X) (34). Cabe señalar que Gajdusek (35) y Lansbury (36) proponen que PrPC y PrPSc están en solución, en equilibrio termodinámico, y que la infectividad viene dada por la formación de agregados multiméricos de PrPSc mediante el reclutamiento de PrPSc monomérico.
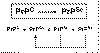
Fig. 1. La reacción en cadena de
los priones.
En cuanto a la conformación tridimensional de PrPC, consta de una mitad carboxi-terminal que, con su tres hélices alfa y una lámina beta, da lugar a una estructura globular estable. La mitad amino-terminal, en cambio, es una cola altamente flexible y en ella tiene lugar el cambio conformacional durante la conversión a PrPSc, consistente en la formación de dominios en lámina beta. De esta manera, mientras que en las preparaciones de PrPC la proporción de hélices alfa es del 43% y la de láminas beta tan sólo del 3%, en las de PrPSc las hélices alfa son responsables del 34% y las láminas beta del 43%.
Los cambios que tienen lugar en la cola amino-terminal son responsables de la resistencia a la degradación que caracteriza a PrPSc y de que la sensibilidad a la proteinasa K propia de la PrPC se torne en resistencia en el caso de la PrPSc. Estas propiedades son aprovechadas por las técnicas que, como el western blot, buscan la distinción entre PrPC y PrPSc. Dependiendo de la sensibilidad o la resistencia de PrP a la proteinasa K, la aplicación de la misma resulta en la eliminación de un número diferente de aminoácidos del extremo amino-terminal de PrP y, por lo tanto, en diferentes mobilidades electroforéticas. La resistencia a la proteinasa K propia de PrPSc también es útil en la demostración inmunohistoquímica de los depósitos de PrPSc (5-8,23,24).
El gen de los priones
Charles Weissmann clonó el gen encargado de codificar tanto la PrPC como la PrPSc (conocido gen Prnp en el ratón y gen PRNP en el humano) y dio un gran impulso a la hipótesis de los priones cuando produjo ratones desprovistos del gen Prnp (ratones knockout) a los que era imposible transmitir la enfermedad mediante la exposición a PrPSc (37-39). En efecto, tal como predica la hipótesis de los priones, la acumulación de PrPSc depende de la disponibilidad de PrPC. Si el ratón no posee el gen Prnp (Prnp-/-), no hay PrPC disponible y la enfermedad no se desarrolla. Lo que es más, si el ratón tiene un solo gen Prnp activo (Prnp+/-) y, por lo tanto, la mitad de PrPC que un ratón normal, la exposición a PrPSc es capaz de provocar la enfermedad, pero ésta se desarrolla mucho más lentamente. Puesto que los ratones libres de PrP son sanos y resistentes a las EET, se ha sugerido la posibilidad de generar ovejas y vacas libres de priones, aunque las dificultades prácticas de tal empresa son fáciles de imaginar (7).
También fue muy importante la demostración de la relación entre las formas familiares de EET y las mutaciones en el gen de los priones, de modo que en todos los casos de ECJ familiar existe alguna mutación en el marco de lectura del gen PRNP. Este gen presenta un polimorfismo en el codón 129, que puede codificar metionina o valina. El 80% de los individuos con ECJ esporádica y el 100% de los enfermos con la vECJ son homocigotos para la metionina en este codón. La heterocigosidad, en cambio, parece conferir cierta protección. Además del gen PRNP probablemente hay otros que influencian la susceptibilidad a las enfermedades priónicas, pero en la actualidad se deconocen. Puesto que la inmunodeficiencia protege contra el scrapie, se ha pensado que algunos de tales genes podrían estar implicados en la maduración de las células linfoides. Sean cuales fueren estos genes modificadores, su influencia sobre la susceptibilidad a las enfermedades priónicas debe ser muy importante, ya que ninguna de las personas que han contraído la vECJ tienen antecedentes ocupacionales de exposición a los priones de la EEB (8).
La barrera entre especies
El uso de ratones knockout (sin gen Prnp) y de ratones transgénicos (con un gen Prnp que no es el suyo propio) ha permitido hacer experimentos que han resultado muy esclarecedores del mecanismo de acción de los priones. Un aspecto particularmente intrigante de la biología de los priones es la existencia de una barrera que limita la propagación de la infección entre diferentes especies. La transmisión de la EEB a los humanos ha suscitado un gran interés sobre los factores que influencian esta barrera entre especies.
Los estudios en ratones transgénicos identificaron la secuencia primaria de PrPC como el determinante principal de la barrera, de modo que las pequeñas variaciones existentes en la estructura primaria de los priones de las diferentes especies protegería a un animal de los priones de otras especies. Pero los priones de la EEB parecen hacer caso omiso de las variaciones secuenciales que puedan presentar los priones de otras especies animales. Esta promiscuidad de los priones de la EEB también provoca inquietud en cuanto a la posibilidad de que la EEB se transmita de las vacas a las ovejas y de éstas a los humanos (8,30,39-41).
Relación entre la EEB y la vECJ
Llegados a este punto, conviene hacer unos comentarios sobre los datos que avalan la transmisibilidad de la EEB a los humanos. En primer lugar, mediante la inyección de homogeneizados cerebrales de vacas con EEB a macacos se han obtenido en éstos lesiones cerebrales muy similares a las que presentan los pacientes con vECJ. Por otra parte, los western blots de extractos cerebrales ponen de manifiesto que los patrones de migración de la EEB y la vECJ son los mismos. Estos patrones de migración son un reflejo del grado de glicosilación del prión y, en tanto que la EEB y la vECJ presentan un patrón de tipo 4 (alto grado de glicosilación), la ECJ esporádica se caracteriza por un patrón de tipo 1 y la ECJ yatrógena por un patrón de tipo 3. El patrón de tipo 4 también es compartido por los extractos cerebrales de los animales de zoológico y los gatos domésticos que han contraído la EEB mediante el consumo de harinas cárnicas y por los macacos a los que se ha transmitido la EEB de forma experimental (8,30,42,43).
Las cepas del prión
Lo dicho sobre la glicosilación de la proteína priónica demanda unos comentarios sobre las denominadas cepas de los priones. Cuando inicialmente se inyectaron a cabras homogeneizados de cerebro de ovejas con tembladera, se comprobó que mientras unas carbras desarrollaban somnolencia otras manifestaban prurito. Esto hizo pensar que una misma proteína priónica tienen diferentes cepas. En la actualidad se han identificado hasta unas 20 cepas distintas del agente del scrapie. Estas cepas, al ser inyectadas a ratones, provocan encefalopatías espongiformes con diferencias en cuanto a los períodos de incubación y a la distribución topográfica y gravedad de los hallazgos neuropatológicos. Además, mediante el western blot puede comprobarse que, debido a sus diversos grados de glicosilación, presentan diferente mobilidad electroforética. Es decir, una misma cadena polipeptídica (una misma estructura primaria) puede dar lugar a una diversidad de cepas cada una de las cuales se caracteriza por una diferente conformación de PrPSc. Cada cepa de PrPSc tendría preferencia por aquellas células nerviosas cuya PrPC presenta un patrón de glicosilación similar al del agente infeccioso (7,43).
HALLAZGOS NEUROPATOLOGICOS
ECJ, kuru, GSS e IFF
El examen macroscópico del encéfalo en la ECJ puede poner de manifiesto hallazgos muy poco llamativos o, dependiendo del tiempo de evolución, signos inespecíficos de atrofia cortical y dilatación ventricular. El cuadro histológico, en cambio, es muy distintivo. Así, durante mucho tiempo, los cambios espongiformes (fig. 2) de la corteza cerebral, ganglios basales, tálamo, corteza cerebelosa y subiculum han sido la piedra angular del diagnóstico de la ECJ. Mediante el estudio ultraestructural puede comprobarse que las vacuolas están rodeadas por una membrana y contienen restos membranosos (fig. 3). Estos cambios espongiformes parecen ser el resultado de la dilatación del retículo endoplásmico y el aparato de Golgi en las dendritas neuronales. En otras enfermedades en las que también tiene lugar una extensa pérdida neuronal, tales como la enfermedad de Alzheimer y la enfermedad de los cuerpos de Lewy, pueden producirse cambios de vacuolización inespecífica del neurópilo que resulten indistinguibles de los cambios espongiformes de la ECJ. Cabe resaltar, por otra parte, que los cambios espongiformes pueden estar ausentes en algunas EET, particularmente en el IFF. Otros hallazgos histológicos de las EET son la pérdida neuronal y la hiperplasia e hipertrofia de los astrocitos y la microglia (fig. 4).
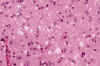
Fig. 2. Cambios espongiformes y
pérdida neuronal en la ECJ (Hematoxilina-eosina).
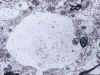
Fig. 3. La microscopia electrónica
permite comprobar que las vacuolas están limitadas por una membrana y contienen
restos membranosos.
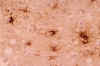
Fig. 4. La inmunotinción para
proteína glial fibrilar ácida pone de manifiesto la astrocitosis reactiva de
la ECJ.
La presencia de abundantes placas amiloideas de PrP viene a completar el cuadro histológico del kuru, aunque el GSS y hasta un 15% de los casos de ECJ también presentan placas amiloideas. Estas placas amiloideas son muy llamativas en el kuru (placas de kuru), en el que afectan sobre todo a la capa granular del cerebelo y presentan un centro eosinófilo del que emergen unas fibrillas radiadas, alrededor de las cuales se dibuja un halo claro. Las placas amiloideas visibles en algunos casos de ECJ también son placas de tipo kuru, mientras que las placas que acompañan al GSS, que tienen una gran predilección por el cerebelo, suelen ofrecer un aspecto multicéntrico, es decir, son el resultado de la agrupación de varios depósitos pequeños. En el IFF, por el contrario, las placas amiloideas están ausentes, siendo el rasgo más notable la pérdida severa de neuronas en el tálamo y las olivas inferiores, algo menos en el cerebelo y prácticamente nada en el córtex cerebral. Esta pérdida neuronal se acompaña de gliosis reactiva, pero los cambios espongiformes son muy poco llamativos o están completamente ausentes (2,18,33,44,45).
vECJ
En la vECJ la vacuolación de las neuronas y el neurópilo, pérdida neuronal y gliosis se acompañan siempre de un hallazgo distintivo consistente en que las placas amiloideas se rodean de un halo de cambios espongiformes («placas floridas») tanto en la corteza cerebral como en la cerebelosa. Los cambios espongiformes son muy llamativos en los ganglios basales y, de forma focal, en la capa molecular del cerebelo, mientras que la gliosis es particularmente prominente en el tálamo posterior. La inmunohistoquímica pone de manifiesto depósitos de PrP en las placas, paredes de los vasos sanguíneos y neuronas pequeñas de las cortezas cerebral y cerebelosa. Además, se obtiene inmunorreactividad para PrP en tejidos linfoides tal como las amígdalas, el bazo y los ganglios linfáticos. Estos hallazgos contrastan con los de la forma esporádica de la ECJ, en la que la gliosis talámica es rara, las placas amiloideas con PrP están presentes en menos del 15% de los casos y no se detecta PrP en los tejidos linfoides (4,46).
Scrapie y EEB
El scrapie se caracteriza por vacuolación que afecta especialmente las neuronas y menos al neurópilo, pérdida neuronal, gliosis, pocas placas amiloideas, cambios severos en los núcleos del tronco cerebral (vestibular, pontinos, olivas, núcleo rojo), columna de Clarke, cerebelo e hipotálamo, sin que haya afectación de la corteza cerebral, el tálamo o el estriado. En cambio, en la EEB hay vacuolación tanto de las neuronas como del neurópilo, pérdida neuronal, gliosis y algunas placas amiloideas, siendo los cambios más severos en el tronco del encéfalo (núcleos del trigémino, núcleo solitario), algo menos en el diencéfalo y mínimos en la corteza cerebral (11,28).
INMUNOHISTOQUIMICA, WESTERN BLOT Y OTROS ESTUDIOS
El estudio histológico convencional de las EET se ha de completar con la demostración de la acumulación de PrPSc en el tejido nervioso, lo que es posible gracias a la disponibilidad de anticuerpos dirigidos contra PrP que permiten la aplicación de técnicas de inmunohistoquímica y western blot. La demostración inmunohistoquímica de los agregados de PrP en tejido parafinado aprovecha la resistencia a la proteinasa K propia de PrPSc, pues si bien los anticuerpos disponibles parecen ser incapaces de distinguir entre PrPC y PrPSc, el pretratamiento con proteinasa K elimina los epítopos amino-terminales en el caso de PrPC y los anticuerpos correspondientes tan sólo proporcionan una inmunorreacción positiva en presencia de PrPSc. KG9, 3F4, 6H4 e IA8 son las clonas de algunos de los anticuerpos comúnmente utilizados para la identificación de PrP.
En la forma esporádica de la ECJ el patrón de inmunotinción de PrP se caracteriza por una positividad puntiforme en el neurópilo y alrededor de los cuerpos neuronales y por la presencia, más infrecuente, de masas globulares extracelulares que se localizan en la vecindad de los cambios espongiformes (fig. 5). La vECJ, en cambio, pone de manifiesto una fuerte inmunotinción en las «placas floridas», así como en numerosas placas de más pequeño tamaño a menudo dispuestas en agregados irregulares (46-48).
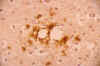
Fig. 5. Evidencia
inmunohistoquímica de depósitos de PrP en relación con los cambios
espongiformes en la ECJ.
El western blot de homogeneizados cerebrales pretratados con proteinasa K proporciona diferentes patrones de bandas de priones diglicosilados, monoglicosilados o no glicosilados que se asocian a la ECJ esporádica (patrón 1), a la ECJ yatrógena (patrón 3) o a la vECJ y la EEB (patrón 4). El patrón de glicoformas de la vECJ, sin embargo, es compartido por el GSS y el IFF (49-51). En cualquier caso, la interpretación de la significación diagnóstica de los patrones del western blot es compleja y es obvio que éste es un método a manejar en centros de referencia, con los que debería consultarse cualquier caso sospechoso de vECJ. En el caso del GSS, el IFF y las formas heredadas de ECJ, la secuenciación del gen PRNP es, por supuesto, un aspecto crucial del estudio.
Comentario aparte merece la biopsia amigdalar para el diagnóstico de la vECJ. En efecto, la presencia de PrPSc en el sistema linforreticular puede utilizarse como uno de los criterios diagnósticos de la vECJ, pero es preciso resaltar que en los órganos linfoides tanto la concentración total de PrPSc como el número de células que ponen de manifiesto la acumulación son muy inferiores a lo que ocurre en el sistema nervioso, por lo que se requiere un alto grado de virtuosismo en la inmunohistoquímica y de sensibilidad en el western blot (46).
Por último, cabe mencionar que la constatación de que el plasminógeno se une selectivamente a PrPSc (y no a PrPC) podría ser de gran utilidad diagnóstica, pues esta propiedad permitiría la detección de PrPSc posiblemente incluso en la sangre periférica. Esta afinidad podría tener también significación patogénica, por cuanto PrPSc, al unirse al plasminógeno, le impediría llevar a cabo la proteólisis de la plasmina, de gran importancia en la remodelación sináptica (52).
INSTRUCCIONES PARA EL MANEJO DE LAS EET
Transmisión y contagio
Las EET no son contagiosas en el sentido habitual del término. Para lograr la transmisión se necesita material específico (sistema nervioso de un individuo afectado) y contacto penetrante. Los estudios epidemiológicos no han identificado ningún factor especial de riesgo para la ECJ esporádica y los casos ocasionales de ECJ habidos en histotécnicos y patólogos (ninguno implicado en estudios de las EET) no parecen constituir evidencia de riesgo por exposición ocupacional a pacientes o tejidos en el medio sanitario. De hecho, el riesgo de infección para el personal anitario se considera muy inferior al que plantean las autopsias de pacientes con hepatitis vírica o infección por VIH, por lo que las precauciones que se toman para éstas son más que suficientes en las necropsias de los enfermos con EET. La confirmación del diagnóstico de EET reviste una importancia obvia y, salvo en las formas familiares en las que basta con el estudio de linfocitos de sangre periférica, requiere la obtención de tejido del sistema nervioso central (53-60).
Biopsias y autopsias
Por lo que se refiere a las biopsias, cuya práctica se desaconseja en los casos típicos, es aconsejable la fijación del tejido en ácido fórmico concentrado durante 1 hora, seguido de fijación en formol durante un mínimo de 48 horas. Alternativamente, la fijación puede hacerse en una mezcla a partes iguales de hipoclorito sódico al 0,1% (lejía casera) y formol durante al menos 48 horas. Tras ello, se lleva a cabo el proceamiento habitual de inclusión en parafina.
En cuanto a las autopsias, es preciso evitar muy cuidadosamente las heridas penetrantes (bisturís de punta roma, guantes de seguridad bajo los guantes convencionales) y protegerse de los aerosoles mediante el uso de una sierra dotada de un sistema de aspiración o, en su defecto, una sierra manual. La mesa de autopsias se cubrirá con un plástico desechable y bajo la cabeza se colocrá una capa gruesa de hojas de papel de filtro para absorber el líquido cefalorraquídeo y la sangre que puedan derramarse. Será suficiente la realización de una autopsia parcial, limitada a la extracción del encéfalo, antes de la cual es aconsejable la obtención de dos muestras en fresco (lóbulo frontal, cerebelo), que se congelarán en nitrógeno líquido y se mantendrán en un congelador a –80°C (para la ulterior realización de estudios de western blots y otros). Si se decide tomar muestras de otros tejidos, conviene hacerlo sin extraer los órganos de las cavidades corporales. Una autopsia completa, con extracción de las vísceras y la medula espinal, ha de llevarse a cabo en una sala de alto riesgo. Se recomienda evitar el embalsamamiento de estos cadáveres. Al finalizar la autopsia, el plástico y el papel de filtro empleados deberán colocarse en un contenedor de residuos sanitarios (cubo negro de los residuos sanitarios del grupo III).
El encéfalo deberá fijarse en formol durante al menos 3 semanas. Las muestras de tejido (menos de 5 mm de grosor) habrán de sumergirse en ácido fórmico concentrado durante un mínimo de 3 horas o en una mezcla a partes iguales de hipoclorito sódico al 0,1% (lejía casera) y formol durante también al menos 3 horas. A partir de este punto se considera que el tejido está descontaminado y se procede a la inclusión habitual en parafina.
Del encéfalo ya fijado conviene tomar muestras de varias áreas del neocórtex cerebral, substancia blanca cerebral, hipocampo, núcleos de la base, tálamos, tres niveles del tronco del encéfalo y varias áreas del cerebelo. Las tinciones imprescindibles son la hematoxilina-eosina, el PAS y el rojo Congo, en tanto que el estudio inmunohistoquímico, además de la demostración de PrP, debe incluir también la detección de amiloide beta y proteína glial fibrilar ácida (53,54,56-60).
Limpieza y residuos
Para la limpieza de los instrumentos puede utilizarse el autoclave (60 minutos a 134°C) o la inmersión en hipoclorito sódico o hidróxido sódico 2N (80 gramos por litro) durante 3 horas. Se aconseja el uso de instrumentos desechables cuando ello sea posible. Por lo que se refiere a la limpieza de superficies, se puede recurrir al hipoclorito sódico o al hidróxido sódico 2N, aunque este último no debe aplicarse a material de aluminio.
Por último, hay que tener en cuenta que todos los residuos sanitarios potencialmente contaminados por priones habrán de tratarse como residuos del grupo III (cubo negro), siendo la única excepción los tejidos fijados en formol no tratados posteriormente con ácido fórmico y el formol postfijación, que serán clasificados como grupo IV (cubo azul). Todos los líquidos procedentes del paciente se considerarán contaminados, por lo que se tratarán como residuos biológicos del grupo III y nunca se eliminarán por los desagües. Los residuos del grupo III se procesan en el autoclave, en tanto que los del grupo IV se incineran (53,54,56-60).
CONCLUSIÓN
La crisis de las vacas locas ha situado en el ojo del huracán unos procesos cuyo diagnóstico definitivo es en gran medida morfológico. Para los patólogos esto supone el reto de estar en la primera línea del frente contra unas enfermedades alrededor de las cuales se ha generado un pánico colectivo, pero también la oportunidad de ser protagonistas en la respuesta a una patología a la vez antigua y emergente cuya comprensión, una vez más, necesita del poder de la morfología. Es obvia la necesidad de laboratorios neuropatológicos de referencia para el estudio y control de las EET, pero esto no sitúa a las enfermedades priónicas fuera del ámbito de interés del conjunto de los patólogos. Los priones, ignorantes de las fronteras tradicionales de la neuropatología, no limitan el escenario de su patogenia al sistema nervioso central y, por ejemplo, tienen una fase de replicación en las células foliculares dendríticas de los centros germinales antes de que la infección se extienda al sistema nervioso central (61). Concordante con ello es que las predicciones del futuro número de casos de vECJ, impenetrables al análisis de modelos matemáticos, parezcan dependientes de los estudios inmunohistoquímicos de PrP sobre piezas de amigdalectomía y apendicectomía (9), que de objetos de desdén podrían convertirse en bienes muy preciados de los archivos de Anatomía Patológica. En cualquier caso conviene tener presente que en la historia reciente es difícil encontrar un grupo de enfermedades que haya poporcionado más sobresaltos que las EET y no es arriesgado predecir que estos enigmáticos procesos aún nos deparan un cúmulo de sorpresas.
BIBLIOGRAFÍA
Epstein FH. Transmissible spongiform encephalopathies. N Engl J Med 1997; 337: 1821-1828.
Ironside JW. Prion diseases in man. J Pathol 1998; 186: 227-234.
Cruz-Sánchez FF, Ariza A. Enfermedades con acumulación de proteína priónica. En: Neuropathologia. Diagnóstico y Clínica. Cruz-Sánchez FF (ed), pp 561-576, Madrid: Edimsa, 2000.
Van der Valk P. Prion diseases. What will be next? J Clin Pathol 1998; 51: 265-269.
Prusiner SB, Scott MR, DeArmond SJ, Cohen FE. Prion protein biology. Cell 1998; 93: 337-348.
Prusiner SB. Prions. Proc Natl Acad Sci USA 1998; 95: 13363-13383.
Aguzzi A, Weissmann C. Prion research: the next frontiers. Nature 1997; 389: 795-798.
Aguzzi A, Brandner S. The genetics of prions — a contradiction in terms. Lancet 1999; 354 (suppl I): 22-25.
Hilton DA. vCJD — predicting the future. Neuropathol Appl Neurobiol 2000; 26: 405-407.
O’Brien M. Have lessons been learned from the UK bovine spongiform encephalopathy (BSE) epidemic? Int J Epidemiol 2000; 29: 730-733.
Hadlow WJ. Neuropathology and the scrapie-kuru connection. Brain Pathol 1995; 5: 27-31.
Creutzfeldt HG. Über eine eigenartige herdförmige Erkrankung des Zentralnervensystems. Z ges Neurol Psychiat 1920; 57: 1-18.
Jakob A. Über eigenartige Erkrankungen des Zentralnervensystems mit bemerkenswertem anatomischem Befunde (spastische Pseudosklerose-Encephalomyelopathie mit disseminierten Degenerationsherden). Z ges Neurol Psychiat 1921; 64: 147-228.
Jakob A. Über eine der multiplen Sklerose klinisch nahestehende Erkrankung des Zentralnervensystems (spastische Pseudosklerose) mit bemerkenswertem anatomischem Befunde. Medizinische Klinik 1921; 17: 372-376.
Jakob A. Spastische Pseudosklerose. En: Monographien aus dem Gesamtgebiete der Neurologie und Psychiatrie. Die Extrapiramidale Erkrankungen. Foerster O, Wilmanns K (eds), vol 37, pp 215-245, Berlín: Julius Springer, 1923.
Richardson EP, Masters CL. The nosology of Creutzfeldt-Jakob disease and conditions related to the accumulation of PrPCJD in the nervous system. Brain Pathol 1995; 5: 33-41.
Zigas V. Laughing Death: The Untold Story of Kuru. Clifton, Nueva Jersey: Humana Press, 1990.
Klatzo I, Gajdusek DC, Zigas V. Pathology of kuru. Lab Invest 1959; 8: 799-847.
Hadlow WJ. Scrapie and kuru. Lancet 1959; ii: 289-290
Cuillé J, Chelle PL. La maladie dite tremblante du mouton est-elle inocuable? C R Acad Sci 1936; 203: 1552-1554.
Gajdusek DC, Gibbs CJ Jr, Alpers MP. Experimental transmission of a kuru-like syndrome to chimpanzees. Nature 1966; 209: 794-796.
Gibbs CJ Jr, Gajdusek DC, Asher DM, el al. Creutzfeldt-Jakob disease (spongiform encephalopathy): transmission to the chimpanzee. Science 1968; 161: 388-389.
Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science 1982; 216: 136-144.
Prusiner SB. Prion diseases and the BSE crisis. Science 1997; 278: 245-251.
Griffith JS. Self replication and scrapie. Nature 1967; 215: 1043-1044.
Brown P, Preece MA, Will RG. «Friendly fire» in medicine: hormones, homografts, and Creutzfeldt-Jakob disease. Lancet 1992; 340: 24-27.
Lane KL, Brown P, Howel DN, et al. Creutzfeldt-Jakob disease in a pregnant woman with a implanted dura mater graft. Neurosurgery 1994; 34: 737-744.
Wells GAH, Wilesmith JW. The neuropathology and epidemiology of bovine spongiform encephalopathy. Brain Pathol 1995; 5: 91-103.
Will RG, Ironside JW, Zeidler M, et al. A new variant of Creutzfeldt-Jakob disease in the UK. Lancet 1996; 347: 921-925.
Collinge J. Variant Creutzfeldt-Jakob disease. Lancet 1999; 354: 317-323.
Gerstmann J, Sträussler E, Scheinker I. Über eine eigenartige hereditär-familiäre Erkrankung des Zentralnervensystems. Zugleich ein Beitrag zur Frage des vorzeitigen lokalen Alterns. Z ges Neurol Psychiat 1936; 154: 736-762.
Lugaresi E, Medori R, Montagna P, et al. Fatal familia insomnia and dysautonomia with selective degeneration of thalamic nuclei. N Engl J Med 1986; 315: 997-1003.
Gambetti PL, Parchi P, Petersen RB, Chen SG, Lugaresi E. Fatal familia insomnia and familial Creutzfeldt-Jakob disease: clinical, pathological and molecular features. Brain Pathol 1995; 43-51.
Telling GC, Scott M, Mastrianni J, et al. Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interaction of cellular PrP with another protein. Cell 1995; 83: 79-90.
Brown P, Goldfarb LG, Gajdusek DC. The new biology of spongiform encephalopathy: infectious amyloidosis with a genetic twist. Lancet 1991; 33/: 1019-1022.
Jarrett JT, Lansbury PT Jr. Seeding «one-dimensional crystallization» of amyloid: a pathogenic mechanism in Alzheimer’s disease and scrapie? Cell 1993; 73: 1055-1058.
Oesch B, Westaway D, Walchli M, et al. A cellular gene encodes scrapie PrP 27-30 protein. Cell 1985; 40: 735-46.
Basler K, Oesch B, Scott M, et al. Scrapie and cellular PrP isoforms are encoded by the same chromosomal gene. Cell 1986; 46: 417-28.
Büeler HR, Aguzzi A, Sailer A, et al. Mice devoid of PrP are resistant to scrapie. Cell 1993; 73: 1339-47.
De Armond SJ, Prusiner SB. Prion protein transgenes and the neuropathology in prion diseases. Brain Pathol 1995; 5: 77-89.
Telling GC. Prion protein genes and prion diseases: studies in transgenic mice. Neuropathol Appl Neurobiol 2000; 26: 209-220.
Hill AF, Desbruslais M, Joiner S, et al. The same prion strain causes vCJD and BSE. Nature 1997; 389: 448-450.
Bruce ME, Will RG, Ironside JW, et al. Transmissions to mice indicate that «new variant» CJD is caused by the BSE agent. Nature 1997; 389: 498-501.
Budka H, Aguzzi A, Brown P, et al. Neuropathological diagnostic criteria for Creutzfeldt-Jakob disease (CJD) and other human spongiform encephalopathies (prion diseases). Brain Pathol 1995; 5: 459-466.
Ironside JW. Review: Creutzfeldt-Jakob disease. Brain Pathol 1996; 144: 379-388.
Ironside JW, Head MW, Bell JE, McCardle L, Will RG. Laboratory diagnosis of variant Creutzfeldt-Jakob disease. Histopathology 2000; 37: 1-9.
Piccardo P, Safar J, Ceroni M, Gajdusek DC, Gibbs CJ Jr. Immunohistochemical localization of prion protein in spongiform encephalopathies and normal brain tissue. Neurology 1990; 40: 518-522.
Bell JE, Gentleman SM, Ironside JW, et al. Prion protein immunocytochemistry — UK five centre consensus report. Neuropathol Appl Neurobiol 1997; 23: 26-35.
Brown P, Coker-Vann M, Pomeroy K, et al. Diagnosis of Creutzfeldt-Jakob disease by Western blot identification of marker protein in human brain tissue. N Engl J Med 1986; 314: 547-51.
Parchi P, Castellani R, Capellari S, et al. Molecular basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Ann Neurol 1996; 39: 767-78.
Collinge S, Sidle KLC, Meads J, Ironside J, Hill AF. Molecular analysis of prion strain variation and the aetiology of «new variant CJD». Nature 1996; 383: 685-90.
Fischer MB, Roeckl C, Parizek P, Schwarz HP, Aguzzi A. Binding of disease-associated prion protein to plasminogen. Nature 2000; 408: 479-483.
Brown P, Cathala F, Gajdusek DC, LaBauge R, Gibbs CJ. Transmission of Creutzfeldt-Jakob disease from formalin-fixed, paraffin-embedded human brain tissue. N Engl J Med 1986; 315: 1614-1615.
OMS. Public health issues related to animals and human spongiform encephalopathies: Memorandum from a WHO meeting. Bulletin of the WHO 1992; 70: 183-190.
Berger JR, David NJ. Creutzfeldt-Jakob disease in a physician: a review of the disorder in health care workers. Neurology 1993; 43: 205-206.
Advisory Committee on Dangerous Pathogens. Precautions for work with human and animal transmissible spongiform encephalopathies. Londres: HMSO, 1994.
Budka H, Aguzzi A, Brown P, et al. Tissue handling in suspected Creutzfeldt-Jakon disease (CJD) and other human spongiform encephalopathies (prion diseases). Brain Pathol 1995; 5: 319-322.
Ironside JW, Bell JE. The «high-risk» neuropathological autopsy in AIDS and Creutzfeldt-Jakob disease: principles and practice. Neuropathol Appl Neurobiol 1996; 22: 388-393.
Alvarez J, Bermejo F, Cabello A, et al. Enfermedad de Creutzfeldt-Jakob y otras encefalopatías espongiformes transmisibles (enfermedades por priones). Guía de información y recomendaciones. Madrid: Instituto Nacional de la Salud, 1997.
Nos A, Domínguez A, Llorens M, Rams N. Prevenció i control de les encefalopaties espongiformes transmissibles als centres sanitaris. Barcelona: Departament de Sanitat i Seguretat Social, 1999.
Bruce ME, Brown KL, Mabbott NA, Farquhar CF, Jeffrey M. Follicular dendritic cells in TSE pathogenesis. Immunol Today 2000; 21: 442-446.
![]()